

DeepMind y el plegamiento de las proteínas
Desde 1994 y cada dos años se viene celebrando una competición para decidir el software que predice mejor la estructura de proteínas a partir de sus secuencias de aminoácidos.
Esta competición conocida por sus siglas: CASP (Critical Assessment of Protein Structure Prediction) fue el escenario de todo un hito en su decimotercera edición, cuando uno de las organizaciones que participaban, DeepMind, quedó primera y con una amplia diferencia.
DeepMind no se dedicaba a estudiar este problema, no era su campo de estudio, pero decidieron inscribirse y con la ayuda del deep learning lograron resultados asombrosos. Pero fue en la decimocuarta edición de la competición (2020) cuando los miembros de DeepMind, con su AlphaFold 2, demostraron que no tenían rival.
Los propios organizadores de la competición afirmaron que AlphaFold 2 resolvía un problema que llevaba planteado 50 años.
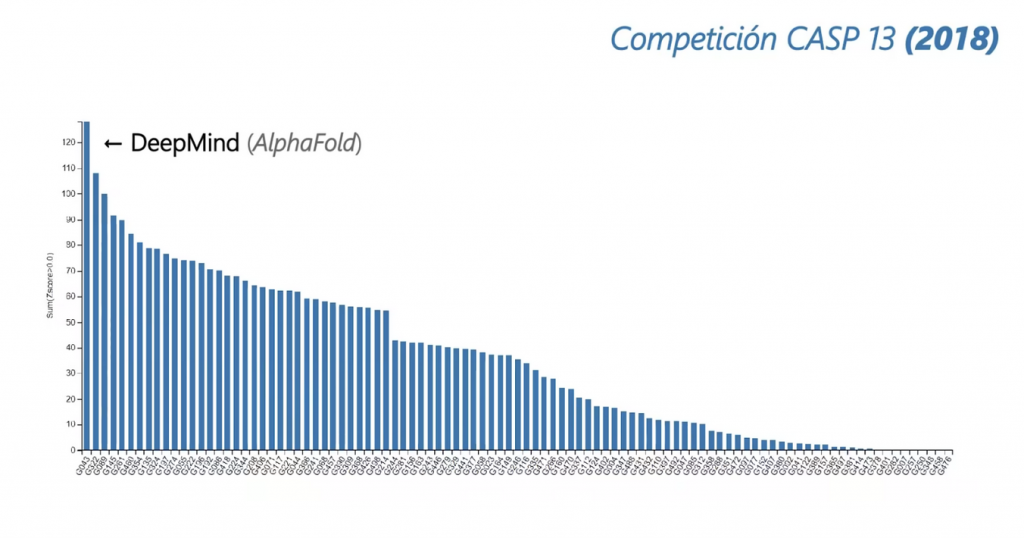
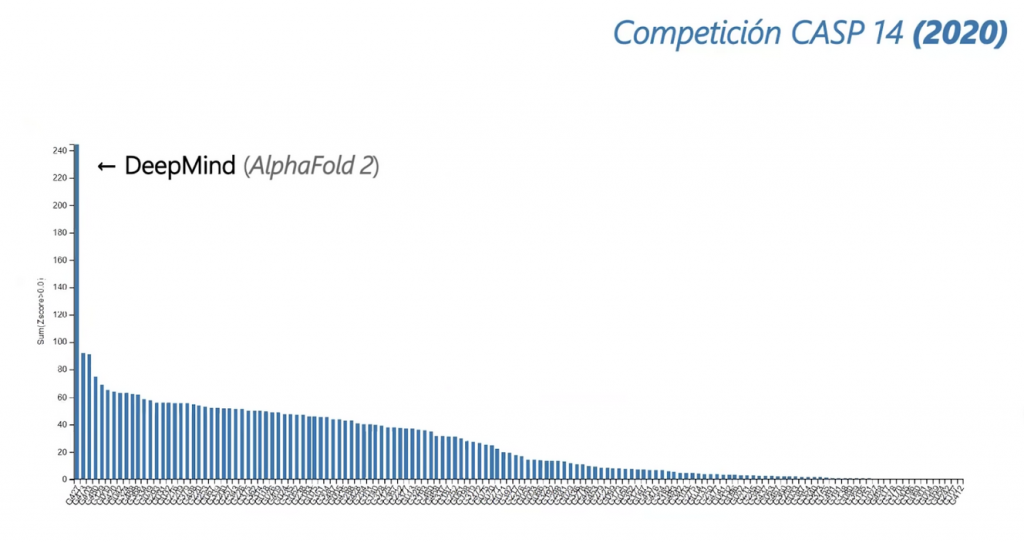
Gráficas extraídas de ¡La Revolución de la INTELIGENCIA ARTIFICIAL en BIOMEDICINA! (AlphaFold2)
Introducción a las proteínas
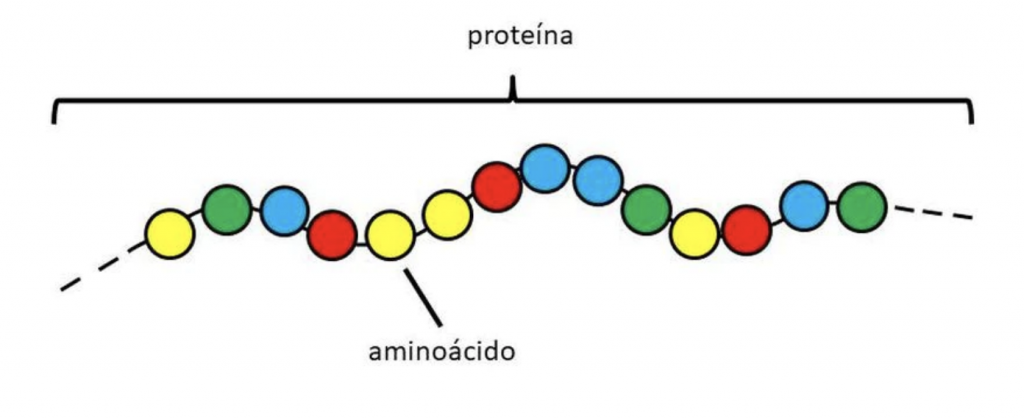
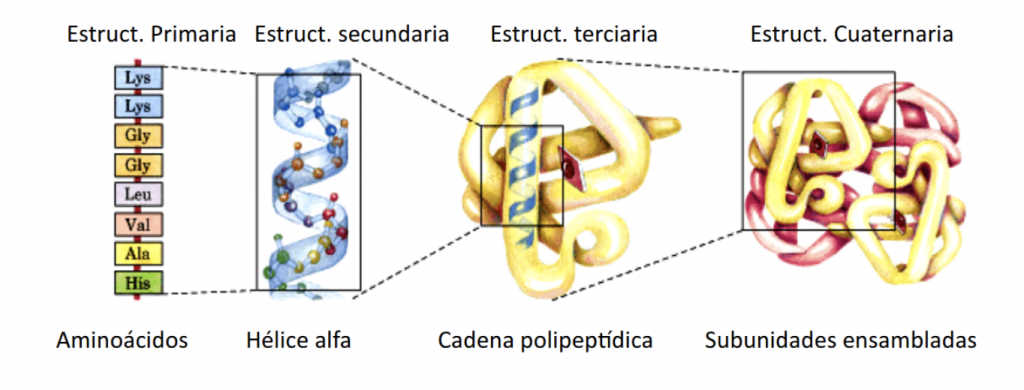
En primer lugar debemos entender que las proteínas son cadenas de longitudes diferentes cuyos eslabones son aminoácidos, pequeñas moléculas formadas por carbono, hidrógeno, oxígeno y nitrógeno, y uno de ellos, la cisteína, también por azufre. La estructura tridimensional de las proteínas depende de su composición, es decir, de los aminoácidos que las constituyen y del orden preciso en que se disponen en la cadena. Esa estructura es muy importante, porque determina su función, pero es muy delicada y factores como el calor, la radiación, o el pH la pueden alterar.
¿Cómo eran las primeras proteínas de la historia?
En la actualidad, para conocer la estructura de las proteínas se utilizan varios métodos. El más tradicional es la cristalografía de rayos X, basada en el análisis del patrón de difracción que se forma cuando estos se dirigen a una sustancia en estado cristalino. El inconveniente de esta técnica es que es laboriosa y no es aplicable a muchas estructuras.
Por otro lado tenemos la criomicroscopía electrónica, más reciente, es una modalidad de microscopía que trabaja con muestras congeladas a temperaturas criogénicas, de manera que se evita la aparición de artefactos.
¿Cómo lo hicieron en DeepMind?
La gran idea de DeepMind fue enfocar el problema de otra forma: en vez de tratar de deducir la estructura tridimensional a partir de su estructura simple y las sus propiedades fisicoquímicas, transformaron este problema en un problema de convertir una imagen a otra imagen y es ahí donde entra el deep learning (aprendizaje profundo) .
En la actualidad tenemos muchos programas y aplicaciones que dada una imagen de entrada nos devuelve una de salida. ¿Quién no ha usado o ha oído hablar de FaceApp? Esa aplicación a la que le cargabamos una foto nuestra y nos devolvía la versión joven, envejecida y de otro sexo de nosotros mismos. Todo gracias a las redes neuronales.
Estas técnicas también se pueden aplicar al sonido. Podemos coger una onda de audio, obtener su espectrograma y con él lograr que la salida sea texto escrito.

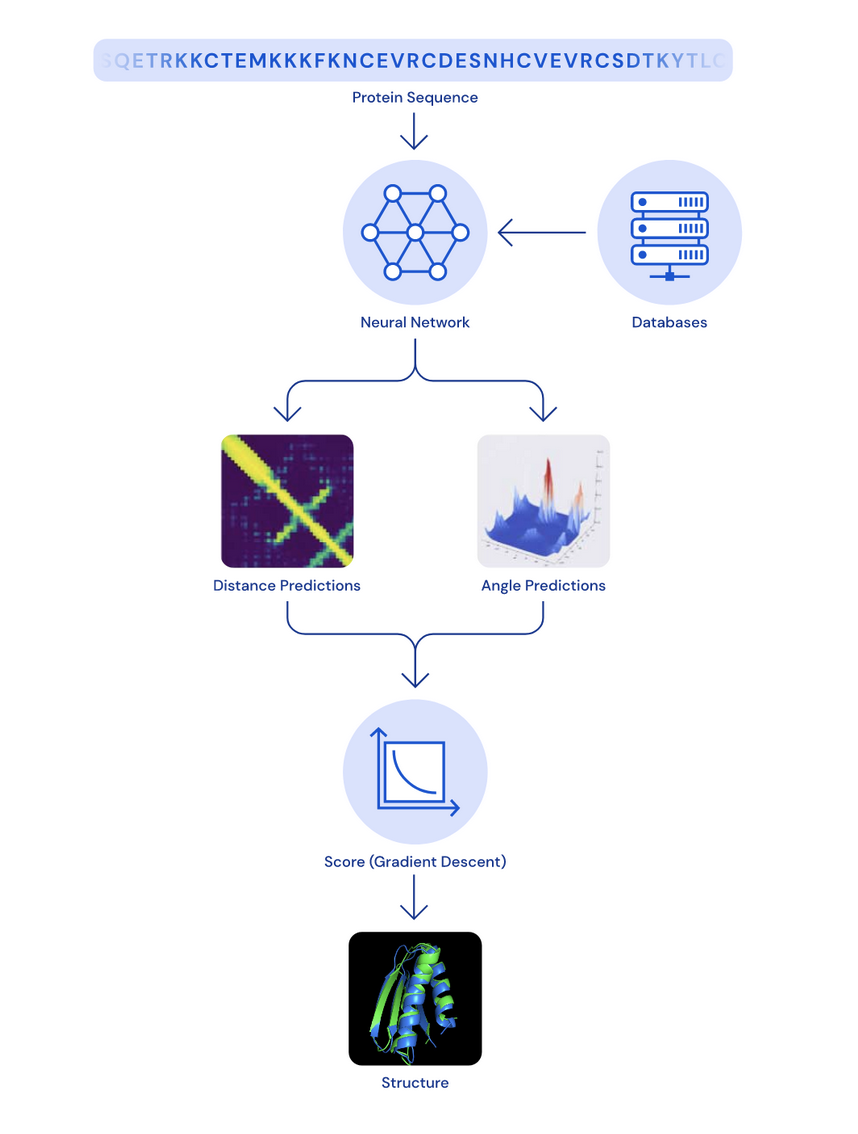
Y eso es lo que hace DeepMind (más o menos), usando técnicas como MSA puede saber qué aminoácidos van a mutar dada una entrada, y luego mediante redes convolucionales (en AlphaFold 1) y transformers (en AlphaFold 2) dada una cadena de aminoácidos logran sacar su matriz de distancias, es decir, una representación matricial de la distancia que hay de un aminoácido a otro en la cadena. A más brillo del píxel, más cerca está ese par de aminoácidos y viceversa.
AlphaFold: Using AI for scientific discovery
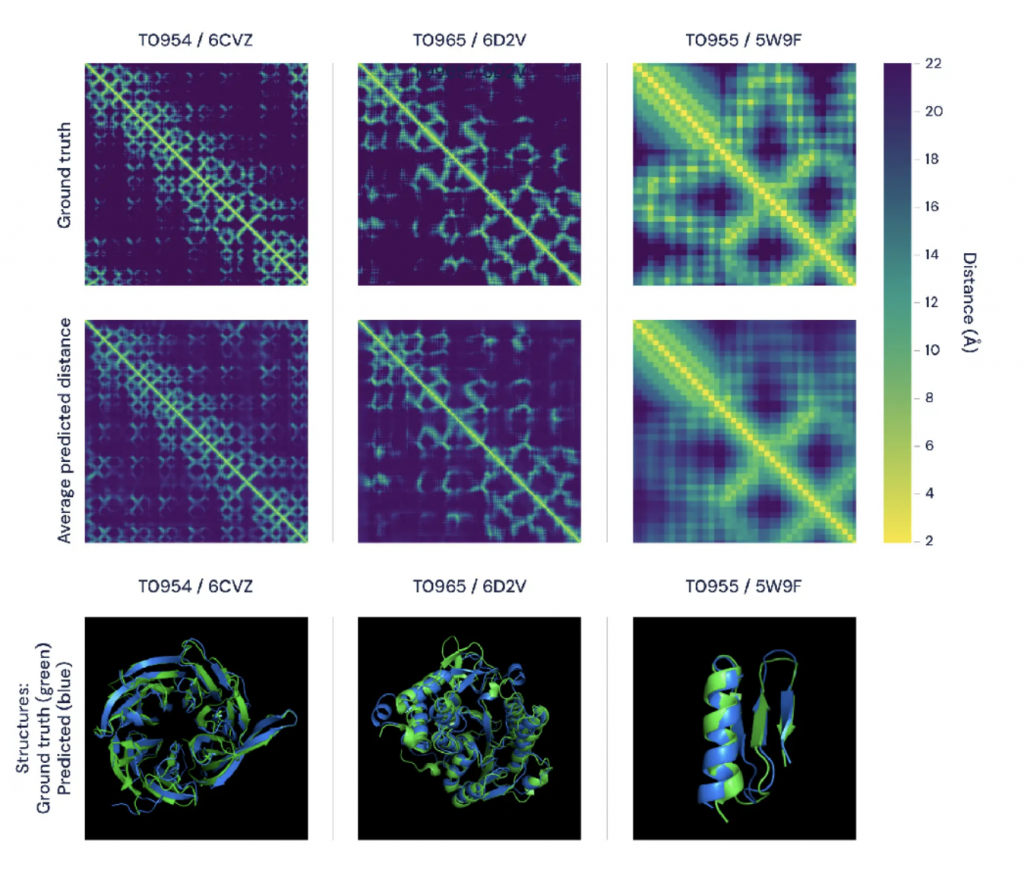
La imagen anterior muestra las matrices de distancia para tres proteínas diferentes. En la fila superior (Ground truth) se muestran las distancias reales determinadas experimentalmente y, en la fila inferior (Average predicted distance), el promedio de las distribuciones de distancia predichas por AlphaFold.
La tercera fila muestra la misma comparación utilizando modelos 3D, con las predicciones de AlphaFold (azul) versus los datos de la primera fila (verde) para las mismas tres proteínas.
¿Pero cómo obtienen esta representación en 3D?, aplicando un algoritmo muy usado dentro del campo del deep learning, el descenso del gradiente (algoritmo de optimización).
Este algoritmo se encarga de optimizar los ángulos de torsión de la proteína hasta que se parezca lo máximo posible a su representación en la matriz de distancias (complejo, lo sé).
A modo de resumen:
Se entrena la red neuronal con la cadena de aminoácidos y una base de datos (hablaremos más adelante de ella), se obtiene una predicción de distancia entre aminoácidos y una predicción del ángulo que forman. Con la ayuda del descenso de gradiente se va ajustando la estructura et voilà, tenemos la estructura tridimensional.
AlphaFold: Using AI for scientific discovery
Entrenamiento del modelo
Ya conocemos a grandes rasgos el proceso que ha hecho DeepMind para que su AlphaFold funcione pero claro, hemos estado hablando de que hay que entrenar las redes neuronales. Para esto han usado aproximadamente 170.000 estructuras almacenadas en el Protein Data Bank junto con grandes bases de datos que contienen secuencias de proteínas de estructura desconocida, de esta forma AlphaFold puede reconocer los patrones que hacen que una proteína se pliegue de una forma u otra.
Recordemos, que es muy importante que todo modelo de inteligencia artificial sea entrenado con datos ya existentes para poder tener un mayor éxito en los resultados. El poder de la inteligencia artificial reside, sobre todo, en los datos que ya tenemos y que tanto nos ha costado conseguir.
Conclusión para el futuro
En palabras del equipo de DeepMind: “AlphaFold es un avance único en una generación, que predice estructuras de proteínas con una velocidad y precisión increíbles. Este salto adelante demuestra cómo los métodos computacionales están preparados para transformar la investigación en biología y son muy prometedores para acelerar el proceso de descubrimiento de fármacos”.
Además han visto indicios de que la predicción de la estructura de las proteínas podría ser útil en futuros esfuerzos de respuesta a una pandemia, como una de las muchas herramientas desarrolladas por la comunidad científica. Han logrado predecir con éxito varias estructuras proteicas del virus SARS-CoV-2, cuyas estructuras se desconocían anteriormente. A pesar de tener muy pocas secuencias relacionadas, han logrado un alto grado de precisión en esas predicciones en comparación con sus estructuras determinadas experimentalmente.
Sin duda alguna estamos ante toda una revolución (otra más) de la mano de la inteligencia artificial. Queda claro que nuestro futuro va a cambiar, o mejor dicho, ya está cambiando gracias a esta gran revolución tecnológica.
Por Alejandro Delgado – Lead Data Scientist en Pixelabs.
Imagen de portada
Share your thoughts
No Comments
Sorry, the comment form is closed at this time.